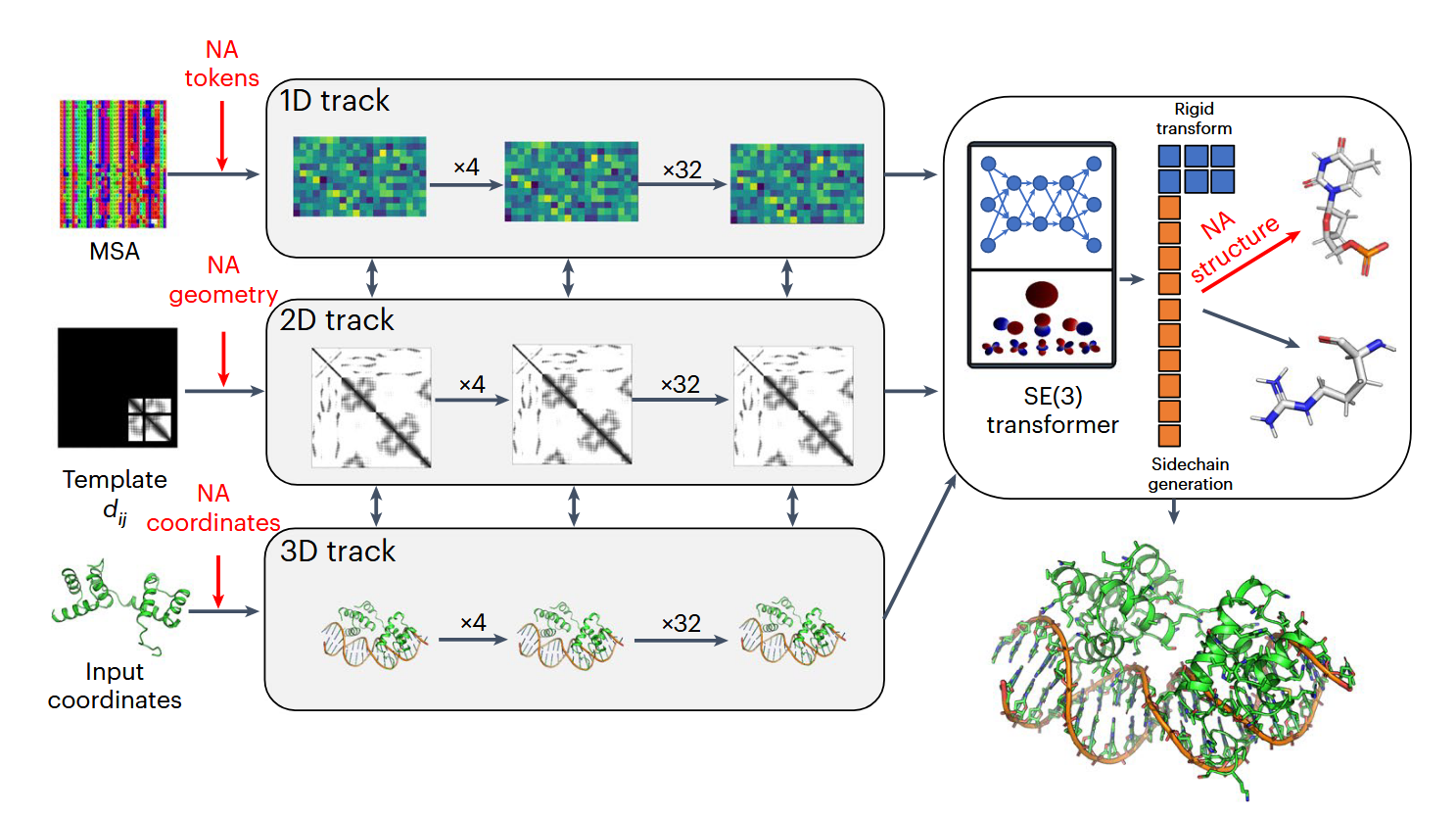
Rose TTAFold NA ( RFNA )的体系结构如图1所示。它基于RoseTTAFold的三轨结构,同时细化了生物分子系统的三种表示:序列( 1D ),残基对距离( 2D )和直角坐标( 3D )。除了几个改进以提高性能外,我们扩展了网络的所有三个轨道,以支持除蛋白质外的核酸。Rose TTAFold中的1D轨道有22个标记,对应于20个氨基酸,一个21:”未知”的氨基酸或间隙标记和一个22:能够进行蛋白质设计的mask标记;为此,我们又增加了10个标记,分别对应于4个DNA核苷酸、4个RNA核苷酸、未知DNA和未知RNA。
图1 | Rose TTAFold NA的体系结构概述。Rose TTAFold NA的三轨结构同时更新了蛋白质-核酸复合物的序列( 1D )、残基对( 2D )和结构( 3D )表示。红色区域强调了核酸掺入所必需的关键变化:1D轨迹的输入包括额外的NA标记,2D轨迹的输入代表模板蛋白- NA和NA – NA的距离(和方向),3D轨迹的输入代表模板或回收的NA坐标。最后,3D轨道和结构精修模块(右上)可以从一个坐标框架(代表磷酸基团)和一组10个扭转角( 6个骨架、3个核糖环和1个核苷)建立全原子核酸模型。在这个图中,\(d_{ij}\)是模板残基间的距离,SE ( 3 )是指三维的特殊欧几里得群。
Rose TTAFold中的二维轨迹建立了一个蛋白质或蛋白质组装体中所有氨基酸对之间相互作用的表示;我们将2D轨道推广到模拟核酸碱基之间以及碱基与氨基酸之间的相互作用。Rose TTAFold中的3D轨道表示每个氨基酸在由3个主链原子( N、CA和C)定义的框架中的位置和方向,最多可以有4个chi ngles来构建侧链。对于Rose TTAFold NA,我们将其扩展为包含每个核苷酸的表示,使用用于描述磷酸基团( P、OP1和OP2)的位置和方向的坐标框架,以及能够建立核苷酸中所有原子的10个扭转角。Rose TTAFoldNA由36个这样的3轨道层组成,之后又增加了4个结构精修层,共计6 700万个参数。
LJ势(Lennard-Jones potential)是一种用于描述分子间相互作用的势能函数,广泛应用于物理和化学领域。LJ势的基本形式包括两个主要部分:吸引力和排斥力。吸引力部分通常表示为 \(u(r)=\frac{A}{r^{12}}\),而排斥力部分表示为\(u(r)=\frac{B}{r^{6}}\),其中 r 是分子间的距离,A 和 B 是常数。
在Rose TTAFold NA不能产生准确预测的情况下,最常见的原因是单个亚基的预测不佳,通常是大的多结构域蛋白、大的RNA ( > 100 nt )和小的单链核酸。当亚基预测准确时,最常见的失效模式是模型要么识别正确的结合取向,要么识别正确的界面残基,而不是两者兼而有之。其余界面完全不正确的情况往往只涉及略过的接触或严重扭曲的DNA。可能是不同的训练计划可以减少这些误差,但更可能是由于这些机制中的训练数据有限。扩展数据图1举例说明。
扩展数据图1 |蛋白质-核酸结构预测的失效模式。( a-d )代表性预测的比较显示,在没有样本集同源物的情况下,预测的常见失效模式。左边是沉积模型,右边是预测模型。( A )个别子单元预测准确性差,导致整体复杂( pdb ID : 6XMF)不正确的例子。这种情况代表了50 %的检查失败,通常是由非常大或非常小的单链核酸( > 100或< 20个核苷酸)、大的多结构域蛋白或严重扭曲的双链DNA造成的。( B )子单元预测精度合理,相对定向正确但界面细节错误( pdb ID : 7A9X)的例子。这样的情况占检查失败的20 %,也可能是由于小的单链核酸或单体结构的轻微偏差造成的。( C )亚基预测准确性高,骨架-骨架结合模式正确,但在DNA ( pdb ID : 4J2X)上的错误位点预测界面的例子。这样的案例占所考察失败案例的10 %。( D )两个亚基预测正确但相对取向和界面不正确的( pdb ID : 7LH9)的例子。这种情况代表了20 %的检查失败,可能是由于扭曲或非双链DNA结构或单体结构的微小偏差造成的。
图3 |多链蛋白质-核酸复合物的建模。a,对161个具有多条蛋白质链或多条核酸链/双链的蛋白质- NA复合物的预测模型精度与实际模型精度的散点图表明,模型准确地估计了误差。b-d,f,训练集中没有同源物的成功预测的例子,显示为沉积模型(左)和预测(右)。其中包括病毒染色质锚定蛋白KSHV LANA ( c , PDB ID : 4uzb)25,两个二聚体螺旋-转角-螺旋转录因子( b , PDB ID : 3u3w ; panel D , PDB ID : 4jcy)26,27和一个复制起始解旋复合物( f , PDB ID : 3vw4)28。E,g,example表示d ( e )和f ( g )所示的相同复合物对同一蛋白质或DNA双链体单独(左)和与另一组分(右)的不同预测构象。
从RFNA的训练集中没有同源物的蛋白质-核酸复合物中,我们选择了8个蛋白质- DNA复合物和6个蛋白质- RNA复合物作为测试案例进行对接。使用AlphaFold5预测蛋白质单体结构,使用相同的MSAs进行RFNA预测,并从模型1 – 5中选择平均预测lDDT最高的预测。使用DeepFoldRNA按照默认的说明进行RNA组分的预测。使用x3DNA生成的DNA双链为B -型螺旋。使用Hdock web server进行对接,仅使用无模板对接,避免直接与原始沉积模型进行拟合。对于RFNA,评估了前3个码头的结构和界面精度。我们承认,更仔细的DNA建模和对接工作流程可以产生更准确的模型,但对于RFNA也是如此。
结合和非结合DNA序列数据集
我们从Cis – BP数据库19中获得了转录因子DNA结合谱的实验数据。我们使用了1509个蛋白质,其中蛋白质序列的实验构建和DNA 8mer E – score是可用的。从每个蛋白质的8mer E – score中,我们选择前3个最富集的DNA序列作为”结合”,3个随机负富集的DNA序列作为”非结合”。我们使用RFNA对蛋白质和DNA进行了联合预测,并根据界面上的平均PAE对模型进行了评估。