用超深度学习模型精确从头预测蛋白质接触图
- 从头预测(De novo prediction)指的是不依赖于同源蛋白质结构模板,而是利用物理学(包括统计物理学)原理根据氨基酸序列进行蛋白质三维结构预测的方法。
- 蛋白质残基接触图(protein contact map):\(L \times L\)的接触矩阵,包含了了解蛋白质结构和功能的关键信息
- 超深度学习模型(Ultra Deep-Learning Model):由两个残差神经网络(ResNet)构成的超神经网络
动机
通过序列来预测蛋白质结构是一个重要的问题,而目前的研究对于缺乏序列同源物的蛋白质接触预测效果很差,因此想通过深度学习方法对蛋白质结构进行从头预测。
方法
通过两个深度残差神经网络形成的超深神经网络,整合进化耦合和序列守恒信息来预测接触。第一个残差网络对序列特征进行一系列的一维卷积变换;第二个残差网络对第一个残差网络的输出、 EC信息和成对电位进行一系列二维卷积变换。通过使用非常深的残差网络,我们可以精确地模拟接触发生模式和复杂的序列结构关系,从而获得更高质量的接触预测,而不管所讨论的蛋白质有多少序列同源物。
结果
十分优异,取得了当时远超其他模型的效果,尤其对于膜蛋白,这个模型十分有效。
作者总结
由于直接进化耦合分析(DCA),蛋白质接触预测和接触辅助折叠已经取得了很好的进展。然而,DCA仅对一些具有大量序列同源物的蛋白质有效。为了进一步改进接触预测,我们借鉴了深度学习的思想,它最近彻底改变了对象识别、语音识别和GO游戏。我们的深度学习方法可以模拟复杂的序列结构关系和高阶相关性(即接触发生模式),从而大大提高接触预测的准确性。
我们的测试结果表明,无论有多少序列同源物可用于所述蛋白质,我们的方法都大大优于最先进的方法。由我们预测的接触指导的从头计算折叠可能比其他接触预测因子折叠更多的测试蛋白。我们的接触辅助3D模型也具有比从训练蛋白构建的同源模型更好的质量,特别是对于膜蛋白。一个有趣的发现是,即使主要使用可溶性蛋白进行训练,本文的方法在膜蛋白上也表现得很好。最近的盲CAMEO测试证实,我们的方法可以用新的折叠和仅少量序列同源物折叠大蛋白。
介绍
一些正确预测的长程接触可以实现精确的拓扑级结构建模,因此,接触预测和接触辅助蛋白质折叠近年来受到了社会的广泛关注。目前的模型对缺少序列同源物的蛋白质接触预测效果差。
- 两个残基形成接触:两个残基在自然结构中空间上接近,即它们的\(c_\beta \) 原子的欧几里德距离小于8Å。
现有的接触预测方法大致分为两类:进化耦合分析(ECA)和有监督机器学习。ECA方法基本需要大量的同源序列,而目前的有监督学习模型基本问题在于网络结构较浅,文章进一步改进有监督学习的方法,通过残差神经网络开发一个大型的深层网络,把蛋白质接触图看作一幅图像,那么蛋白质接触预测有点类似像素级图像标记,因此一些有效的图像标记技术也可以用于接触预测,但有些难点:
- CV领域像素级图像标记的研究过少
- 在许多图像分类场景中,图像大小被调整到一个固定的值,但是接触图的大小不能调整
- 输入特征更加复杂
- 蛋白质接触比例过少(<2%),表示正负标签极不平衡
接触预测的深度学习模型
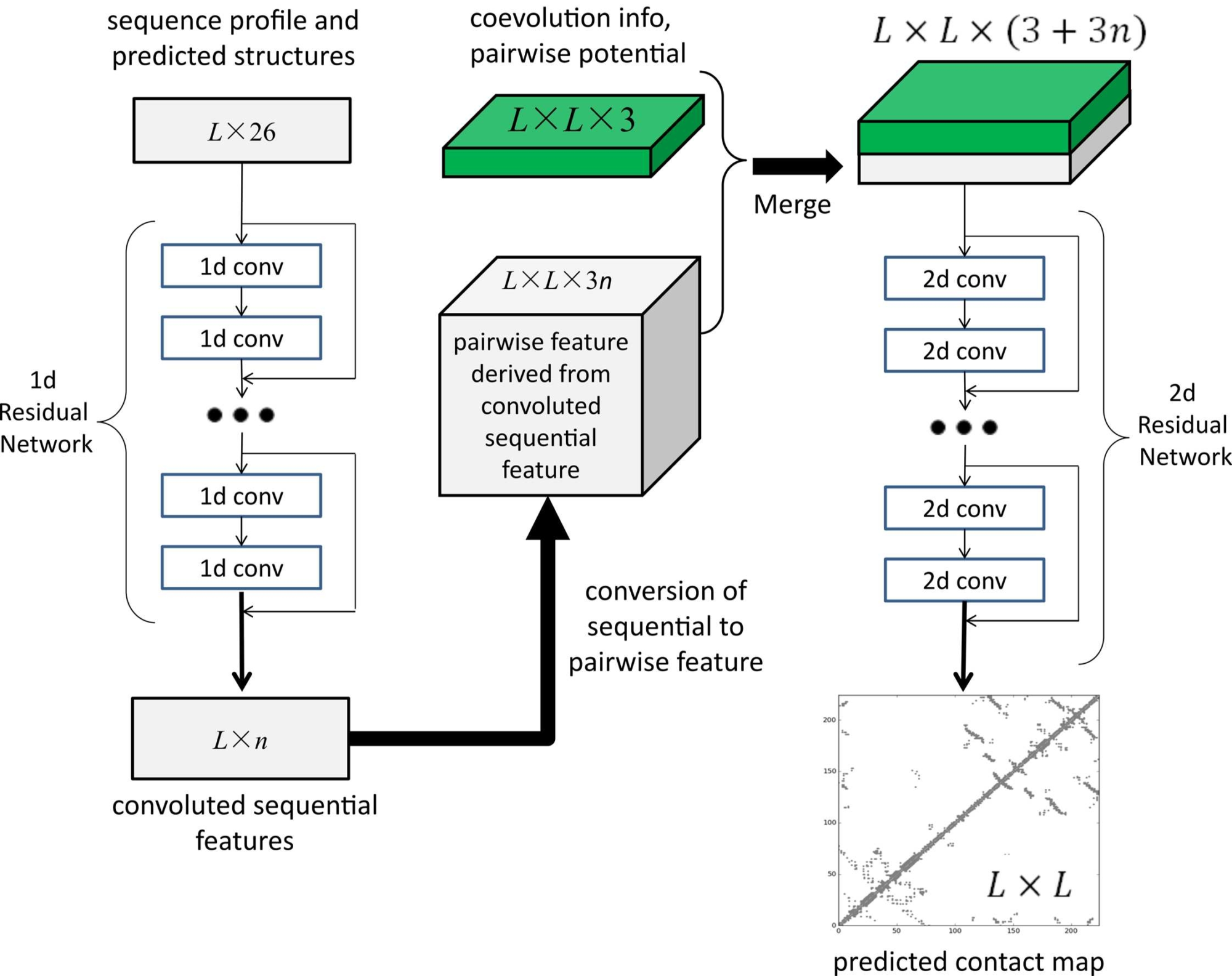
图1说明了本文的接触预测深度神经网络模型。与之前用于接触预测的,仅使用少量隐藏层(即,浅层架构)的监督学习方法不同,本文的深度神经网络使用了数十个隐藏层。通过使用非常深的架构,模型可以自动学习序列信息和接触的复杂关系,并对接触之间的相互依赖性进行建模,从而改进接触预测。

模型由两个主要模块组成,每个模块都是残差神经网络。第一个模块对序列特征(序列谱(sequence profile)、预测的二级结构和溶剂可及性)进行一系列一维(1D)卷积变换。该1D卷积网络的输出通过外部级联(一种类似于外积的操作)转换为二维(2D)矩阵,如图2所示。
从序列特征到成对特征的转变(外部级联) \( (L \times n -> L \times L \times (3n+3))\):假设1D残差网络的输出为\( v=\{v_1,v_2,…,v_L \} \),\(L\)代表着蛋白质序列的长度,vi是一个n维的特征向量,它代表着残差块输出的对应氨基酸残基的特征。对于一对残基\(i\)和\(j\),我们将\(v_i、v_{(i+j)/2}\)和\(v_j\)进行拼接成为一个\(3n\)长度的特征向量,该特征向量就作为残基对的输入特征,最后得到\( L \times L \times 3n\)的特征矩阵。再将使用CCMpred计算得到的EC信息和成对的电位信息作为特征与上述矩阵进行拼接,就得到了2D残差网络的输入特征矩阵,为一个\( L \times L \times (3n+3)\)的矩阵。

然后与成对特征(即共同进化信息和距离电势)一起馈送到第二模块中。第二模块是对其输入进行一系列2D卷积变换的2D残差网络。最后,2D卷积网络的输出被馈送到逻辑回归中,该逻辑回归预测任意两个残差形成接触的概率。此外,每个卷积层之前还进行了一个简单的非线性变换,称为校正线性单元。数学上,1D残差网络的输出只是一个维度为\(L\times m\)的2D矩阵,其中m是网络的最后卷积层生成的新特征(或隐藏神经元)的数量。
生物学上,这个1D残差网络学习残差的顺序上下文。通过堆叠多个卷积层,网络可以在非常大的连续上下文中学习信息。2D卷积层的输出具有维度\(L \times L \times n\),其中\(n\)是该层为一个残差对生成的新特征(或隐藏神经元)的数量。2D残差网络主要学习接触发生模式或高阶残差相关性(即残差对的2D上下文)。每一层隐藏神经元的数量可能不同。
import theano.tensor as T
##一个类似于两个向量的外积的操作,但这里我们做的是级联,而不是一个向量与自身的乘积
##输入的形状(batchSize,seqLen,n_in),输出的形状(batch Size,seq Len,seqLen,2*n_in)
def OuterConcatenate(input):
##an operation similar to the outer product of two vectors, but here we do concatenation instead of product of one vector with itself
##input has a shape (batchSize, seqLen, n_in), output has shape (batchSize, seqLen, seqLen, 2*n_in)
seqLen = input.shape[1]
input2 = input.dimshuffle(1, 0, 2)
x = T.mgrid[0:seqLen, 0:seqLen]
out = input2[x]
output = T.concatenate((out[0], out[1]), axis=3)
return output.dimshuffle(2, 0, 1, 3)
-----------------------------------------实际使用的代码--------------------------------------------
##input has a shape (batchSize, seqLen, n_in)
##this function returns a output with shape (batchSize, seqLen, seqLen, 3*n_in) such as
## output[:,i ,j, :] = input[:, (i+j)/2, :], input[:, i,:], input[:, j,:]
##input具有形状(batchSize,seqLen,n_in)
##此函数返回形状(batch Size,seq Len,seqLen,3*n_in)的输出,例如
##output[:,i ,j, :] = input[:, (i+j)/2, :], input[:, i,:], input[:, j,:]
def MidpointFeature(input, n_in):
seqLen = input.shape[1]
x = T.mgrid[0:seqLen, 0:seqLen]
y1 = x[0]
y2 = (x[0] + x[1]) / 2
y3 = x[1]
input2 = input.dimshuffle(1, 0, 2)
out1 = input2[y1]
out2 = input2[y2]
out3 = input2[y3]
out = T.concatenate([out1, out2, out3], axis=3)
final_out = out.dimshuffle(2, 0, 1, 3)
n_out = 3 * n_in
return final_out, n_out方法
残差模块:残差神经网络由多个残差块构成,主要结构如图,一个残差块由2个卷积层和2个激活层组成,Xl和Xl+1分别是块的输入和输出,激活层使用ReLU函数。 为了加快训练速度,实际网络在每个激活层之前添加了一个批处理规范化层(BN)。1D卷积层使用的滤波器尺寸(即窗口尺寸)为17,而2D卷积层使用的滤波器尺寸为3×3或5×5。通过将许多残差块叠加在一起,即使在每个卷积层使用了一个小窗口大小,网络也可以模拟输入特征和接触之间的非常长的相互依赖关系,以及两个不同残基对之间的长程互依关系。文章将一维残差网络的深度(即卷积层数)固定为6,但改变二维残差网络的深度。实验结果表明,在每个位置有大约60个隐藏神经元,第二个残差网络有大约60个卷积层,模型可以产生很好的性能。此外,具有较小窗口大小的2D卷积神经网络也比具有较大窗口大小的网络具有较少的参数数目,训练参数更少,训练速度更快。

*ResNet为什么效果好:
- 深度网络的效果一般比浅层网络的效果要更好,假设我们已经存在某个K层的网络是最优的网络,那么我们可以构造一个更深的网络,其最后几层仅是该网络第K层输出的恒等映射,就可以取得与一致的结果。但实际上VGG网络深到一定程度,网络实现了退化,不是过拟合,因为深网络的训练误差和测试误差都更低。一个合理的猜测就是,对神经网络来说,恒等映射并不容易拟合。而ResNet解决了这个问题,它的残差模块被设计成一个天然的恒等映射。
- 一定程度解决了深层网络梯度爆炸或消失的问题,残差结构可以使得前后梯度传播更流畅。
损失函数:采用极大似然损失函数对模型参数进行训练,损失函数被定义为训练蛋白所有残基对上的负对数似然平均。由于所有残基对之间的接触比很小,为了使训练算法快速收敛,文章对形成接触的残基对赋予较大的权重。权重的分配使得分配给形成接触的残基对的总权重大约是训练集未形成接触的残基对的1/8。
(使用L2正则并使用随机梯度下降,一般20-30 epoch收敛)
如何处理蛋白质序列长度不同的问题:分小批量进行训练,将蛋白质按照序列长度进行分类,长度相近的分类在同一个批量中,如果同一个批量中的蛋白质序列长度不一致,那么对短的蛋白质序列进行零填充,使其长度相同。而不同的batch之间是可以容忍长度不同的,因为卷积操作和蛋白质的长度没有关系。
训练集和测试集:我们的测试数据包括150个Pfam家族、105个CASP11测试蛋白、76个硬CAMEO测试蛋白(S1表)和398个膜蛋白(S2表)。所有被测膜蛋白的残基长度不超过400个,任意两个膜蛋白的序列同源性均小于40%。对于CASP测试蛋白,我们使用官方的域定义,但是我们不将CAMEO或膜蛋白解析成域。我们的训练集是2015年2月创建的PDB25的一个子集,其中任意两个蛋白质的序列同源性小于25%。如果一个蛋白质满足以下条件之一,我们将其从训练集中排除:(i)序列长度小于26或大于700,(ii)分辨率低于2.5ï,(iii)具有由多个蛋白质链组成的结构域,(iv)没有DSSP信息,以及(v)其PDB、DSSP和ASTRAL序列之间存在不一致性。为了消除测试集的冗余,我们排除了与任何测试蛋白共享>25%序列同源性或BLAST E值<0.1的任何训练蛋白。在我们的训练集中总共有6767个蛋白质,我们从中训练了7个不同的模型。对于每个模型,我们从训练集中随机抽取约6000个蛋白质来训练模型,并使用剩余的蛋白质来验证模型并确定超参数(即正则化因子)。最后的模型是这7个模型的平均值。
蛋白质的序列特征\((L \times 26)\):残差网络的输入特征包括蛋白质序列轮廓、预测的3态二级结构和3态溶剂可及性。
CCMpred生成的EC信息和成对电位用于与第一个残差网络的输出进行合并。
对于一个训练蛋白,我们使用PSI-BLAST(E值0.001和3次迭代)搜索2012年10月的NR(非冗余)蛋白质序列数据库,找到其序列同源序列,然后构建其MSA和序列轮廓,并预测其他特征(即二级结构和溶剂可及性)。(不太懂)序列profile表示为二维矩阵,维数为L×20,其中L为蛋白质长度。预测的二级结构用维数为L×3的二维矩阵表示(每个条目都是一个预测分数或概率),预测的溶剂可达性也是如此,将它们拼接在一起,得到一个维数为L×26的二维矩阵,作为1D残差网络的输入。
评价指标:评估L/k(k=10,5,2,1)预测接触的准确性,其中L是蛋白质序列长度。预测精度定义为在L/k预测值最高的接触中ground-turth的百分比。作者还根据一个接触上两个残基的序列距离将其分为三组。也就是说,当一个触点的序列距离分别为[6,11]、[12,23]和≥24时,它是短距离、中距离和长距离。
结果:文章的方法大大优于现有的方法,可以做更精确的接触辅助折叠。对105个CASP11靶点、76个CAMEO hard靶点和398个膜蛋白进行测试,用文章的方法、一个具有代表性的EC方法CCMpred和CASP11冠军方法 MetaPSICOV得到的平均top L长程预测精度分别是0.47、0.21和0.30;文章的方法、CCMpred和MetaPSICOV的top L/10远程平均精度分别为0.77、0.47和0.59。使用文章预测的接触作为约束条件但没有任何力场的从头算折叠可以为579个测试蛋白质中的203个产生正确的折叠(即TMscore>0.6),而使用MetaPSICOV-和CCMpred预测接触的方法分别只能对其中79个和62个产生正确的折叠。文章的接触辅助模型也比基于模板的模型有更好的质量,特别是对于膜蛋白。根据文章的接触预测建立的3D模型中,有208个膜蛋白的TMscore>0.5,而同源模型中只有10个的TMscore>0.5。此外,即使主要是通过可溶性蛋白质训练,深度学习方法对膜蛋白也非常有效。在17年Blind CAMEO基准测试中,文章实现该方法的全自动web服务器成功地折叠了6个目标,只有0.3L-2.3L的有效序列同源,包括一个182个残基的β蛋白,一个125个残基的α+β蛋白,一个140个残基的α蛋白,一个217个残基的α蛋白,260个残基中有一个α/β,462个残基中有一个α蛋白。